
- ホーム
- 読みもの
- WPIで生まれた研究
- 「次元の呪い」を解いて生物学の研究レベルアップへ(WPI-ASHBi)
「次元の呪い」を解いて生物学の研究レベルアップへ(WPI-ASHBi)
2024年05月08日掲載
今や生物学の研究現場で不可欠のツールとしての地位を獲得したシングルセル遺伝子発現解析。サンプルに含まれる細胞1つ1つについて調べ、全遺伝子のRNA発現量をデータ化する技術だ。基礎研究から臨床まで、その活用法は様々。Science誌が2018年のBreakthrough of the yearの1位にシングルセル遺伝子発現解析(single-cell RNA-sequencing; scRNA-seq)を選出したことからも、その革新性に対する期待の大きさがうかがえる。
これが大きな可能性を秘めた技術であるのは間違いない。しかし課題もある。京都大学ヒト生物学高等研究拠点(WPI-ASHBi)准教授の中村友紀さんによれば、シングルセル遺伝子発現解析の結果が「従来の生物学実験で得られた知見と合わないことがしばしばあった」という。
 WPI-ASHBi准教授 中村さん
WPI-ASHBi准教授 中村さん
シングルセル遺伝子発現データ解析では細胞種の分類や細胞分化経路の追跡が一細胞単位で可能とされる。つまり、どんな遺伝子がどれくらい発現しているかを測定して細胞をグループ分け(クラスタリング)したり(たとえば同じ遺伝子群を発現している細胞なら仲間と評価する)、細胞が分裂をくり返しながら性質を変える様子(細胞分化)を追跡したりすることができると言われるが……。
次元の呪い
「解析結果を細かく見ると、類似性が高いはずの細胞が離れた別のクラスターに分類されたり、あるべき細胞分化の繋がりが切れてしまったりすることがよくあったんです。全体として間違っているわけではないので大目に見ている研究者が多かったのだと思いますが、以前から気になっていました」
この問題意識をきっかけに生まれたのが、2022年8月9日、Life Science Alliance誌で公開された論文「Resolution of the curse of dimensionality in single-cell RNA sequencing data analysis(シングルセル遺伝子発現データ解析における次元の呪いの解決)」だ。
WPI-ASHBiの研究チームは論文で「RECODE(レコード)」と呼ばれるシングルセル遺伝子発現データ解析の前処理法を提案している。これを使うと、これまでクラスタリングや細胞分化経路の追跡で生じていた「欠落」部分が復活するばかりか、細胞種の微妙な違いを区別できるようになるという。
この研究成果は最もWPIらしい論文の一つだ。なぜなら一見かけ離れた生物学と数学の融合の賜物だからだ。
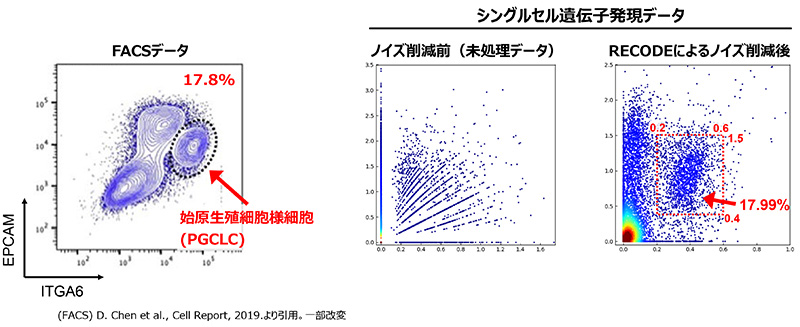 RECODE処理前後の遺伝子発現分布の比較:左はFACS(フローサイトメトリー)による“真実”に近い遺伝子発現分布。中央は未処理のシングルセル遺伝子発現データ、右はRECODE処理後の遺伝子発現分布。 FACSデータでは3つのクラスターが観測され、右上のクラスターが始原生殖細胞様細胞の集団を示す。未処理のシングルセル遺伝子発現データではノイズの影響でクラスターが観測されないのに対し、RECODE処理後のデータはFACS同様のクラスターが形成されている。
RECODE処理前後の遺伝子発現分布の比較:左はFACS(フローサイトメトリー)による“真実”に近い遺伝子発現分布。中央は未処理のシングルセル遺伝子発現データ、右はRECODE処理後の遺伝子発現分布。 FACSデータでは3つのクラスターが観測され、右上のクラスターが始原生殖細胞様細胞の集団を示す。未処理のシングルセル遺伝子発現データではノイズの影響でクラスターが観測されないのに対し、RECODE処理後のデータはFACS同様のクラスターが形成されている。
2019年5月のゴールデンウィーク中、WPI-ASHBi特定准教授(当時は特定助教)の井元佑介さんは、あることに気付いた。中村さんが言っているシングルセル遺伝子発現データ解析の問題は、次元の呪いと関係があるかもしれない——。
次元の呪いとは、データの次元が増えれば増えるほど、データ解析の精度が下がる現象だ。ここで言う次元とは要素数あるいは項目数と言いかえることができる。たとえばある学級の生徒全員の体重を集めたデータは1次元、体重と身長を集めたデータは2次元である。ヒトの場合、各細胞(のゲノム)には約2万の遺伝子が含まれる。つまり次元(項目数)が約2万にも達する。
「シングルセル遺伝子発現解析で得られるのは典型的な高次元データです。この場合、データに含まれるノイズ(誤差)が計算時に蓄積して、本来の生物学的な情報が得られなくなってしまいます」(井元さん)
 WPI-ASHBi特定准教授 井元さん
WPI-ASHBi特定准教授 井元さん
一般的なデータ解析の手法で計算すると、「塵も積もれば山となる」の諺のように、項目ごとのノイズは小さくても、項目が増えれば増えるほどノイズが蓄積してしまい、必要な情報がノイズに埋もれて見出しがたくなるのだ。これに対し従来のシングルセルデータ解析手法では重要と見なした遺伝子や情報のみを選択し、そうでない情報を切り捨てる(次元を削減する)ことでノイズの蓄積問題を回避していた。しかし選択する情報が本当に生物学的に重要かどうかが保証されておらず、さらには捨て去る情報に重要な情報が残されている可能性もあった。
RECODEで呪いを解く
井元さんは九州大学大学院で数理学の学位を取得した後、東北大学を経て、2019年2月にWPI-ASHBiに着任。最初は生物学における問題を理解するのに苦労したという。
「遺伝子という言葉は知っていても、その意味を理解していなかったんです。遺伝子とクロモソーム(染色体)の違いも説明できなかった。中学校で生物を一応学びましたが、その後、触れる機会がないまま10年以上経っていましたから……。今でもよく覚えていますが、WPI-ASHBiに来て初日のセミナーでエクソンとイントロンの説明を聞いても何一つ理解できませんでした(笑)」
途方に暮れつつも生物学の基礎を勉強しつつ、生物学のデータ解析に役立ちそうな専門書にも目を通した。その中の一冊が、高次元統計学を扱った本だった。
「次元の呪いにどう対処するかを考えるのが高次元統計学と言っても過言ではありません。2000年代に入って急速に発展してきた分野です」
通常の統計学では、サンプル数を増やしたときのデータやその統計量の振る舞いを考える。一方、高次元統計学ではサンプル数だけでなく次元も同時に増やしたときに、データがどんな振る舞いをするかを考える。
「サンプル数nと次元dに対して、従来の統計学ではnが増えたらどうなるかについて議論されてきました。それではdを増やしたらどうなるのかを考えるのは数学者なら自然です。日本では筑波大学の青嶋誠さんと矢田和善さんが精力的にこの分野の研究に取り組んでこられた。彼らの論文を調べているうちにシングルセル遺伝子発現解析データには彼らが開発した手法を使えば、ノイズを削減できそうだと気付きました」
一方、井元さんから新しいアイデアを聞かされた中村さんは戸惑ったという。
「scRNA-seqは試料が微量ゆえノイズが大きいのは知っていましたが、そんなに悪影響を及ぼすとは思っていなかったんです。むしろ問題はクラスタリングの仕方にあるのだろうと予想していました。井元さんには『クラスタリングのいい方法を教えて』と話していたんです。ところが井元さんは『次元の呪いじゃないか』って言ってきて。はじめて聞く言葉で、最初は何のことか分かりませんでした(笑)」
かくして本格的な融合研究がスタートすると、わずか2カ月で井元さんはノイズ削減技術の試作版を作りあげた。
「井元さんがあっという間に作ったので驚きましたが、自分が持っていたデータに適用すると良好な結果が得られました。それが後に論文で発表することになるRECODEのバージョン0ですね」(中村さん)
井元さんは中村さんのフィードバックを受けながらノイズ削減技術の改良を重ねた。一方、研究の対象であるシングルセル遺伝子発現解析にも新たな動きが見られた。それまで解析法が乱立していたが、10X Genomics社のChromiumシリーズが優勢になってきたのだ。
「今でも適材適所で他のシングルセル技術も使われていますが、10Xが広く普及しています」(中村さん)
10X社のscRNA-seq法は、細胞から取り出すmRNAにUMI(Unique Molecular Identifier)と呼ばれる分子バーコードを付加できる仕組みを取り入れていた。mRNAの情報はcDNAに置きかえられ、PCR(ポリメラーゼ連鎖反応)により増幅してNGS(次世代シーケンサー)で解読という流れでシングルセル遺伝子発現データの取得は進む。PCRの増幅率は一定でないため、データにノイズが混じるが、これを除くのがUMIである。
UMIですべてのノイズが回避できるならRECODEも不要だが、話はそれほど簡単ではない。どうしても回避できないのが、mRNAからcDNAへの逆転写時とcDNA断片を採取するときに生じるノイズだ。一細胞から取り出す全mRNAのうち平均して10%未満しか検出できないことに起因し、ランダムサンプリングノイズと呼ばれる。
厄介なのは、検出率の「平均」なので、細胞Aから10%検出する場合もあれば、細胞Bからは8%検出する場合も、細胞Cからは12%検出する場合もあるといった事態を想定しなければならないことだ。実際のシングルセル遺伝子発現データでは、細胞の状態の違いを表す生物学的なばらつきと、このランダムサンプリングによる非生物学的なばらつきがミックスして観測される。特に発現量が小さい遺伝子のmRNA量のばらつきや、希少細胞種のような少数派のばらつきがランダムサンプリングノイズによって隠されてしまうのだ。
退治すべきノイズの性質がはっきりしたので、井元さんは遺伝子発現データのランダムサンプリングノイズを数学的にモデル化し、先に触れた筑波大の矢田氏・青嶋氏が開発した高次元統計学的な手法を応用できる形に置きかえる新たな正規化手法(noise variance-stabilizing normalization:ノイズ分散安定正規化)を考案。かくしてRECODEが完成したのは2020年2月頃のことだった。
実はRECODE以前にもシングルセル遺伝子発現データのノイズ自体に対処する手法も非常に多く提案されてきた。しかし、従来のノイズ削減手法はノイズの分布を正しくモデル化できておらず、細胞が持つ真のばらつきまで操作してしまう。そのため、ある解析結果の再現性を向上させる一方、別の解析結果の再現性を下げる、つまりあちら立てればこちらが立たぬ短所があった。それに対してRECODEはノイズの分布を正確にモデル化し、ノイズのみをデータから削減するので、全体的にデータ解析の精度が上がった。

生物学の研究水準向上へ
井元さん、中村さんにとってRECODEの従来手法に対する優位性は明らかだった。しかし研究成果をまとめた論文の査読者への対応には苦労した。
「本研究成果はバイオインフォマティクス学的研究に位置づけられるので、しかるべき投稿先を選んだのですが、私の専門は数学、中村さんの専門は生物学で、どちらもバイオインフォマティクスの研究者なら身につけているはずの『お作法』を知らない。査読者から『我々の世界ではこういうデータを使うのが当たり前だ』と言われ、ほぼ全面的に書き直しになったこともある。関連する学会に出て助言を求めようにも、コロナ禍でそういう機会を得るのが難しく、きつかった」(井元さん)
中村さんはRECODEの威力を示すデータを探すのに苦労したという。
「RECODEの正しさの証明には、他の実験手法などにより正解が明らかになっているデータが必要です。たとえば皮膚細胞と血液細胞を混ぜてシングルセル解析すれば違いは明らかなので、解析結果が正しいかどうかを示すことができます。しかし元々違いの大きい細胞同士なので、わざわざRECODEの前処理でノイズ削減しなくてもきれいなクラスターに分けることができてしまいます。そうかといって、生物学的に近い細胞同士の場合、RECODEを使ってもクラスターに分けられませんが、元々似ているので分けられなくて当然です。したがって生物学的な実験で分けられることがすでに明らかでありながらノイズのせいできれいに分けられないデータを見つける必要がありました」
そのような絶妙なデータの一つとして、マウスの原腸陥入期の胚の解析結果が論文で紹介されている。原腸陥入とは、胚の表面の一部が凹んで内側に入りこむ(最終的には肛門から口まで貫通する)ダイナミックな形態形成運動である。これによって外胚葉(後に神経、外皮などになる)、中胚葉(後に骨、筋肉などになる)、内胚葉(後に肺や消化器になる)が形成される。
「過去の研究から、消化管になる内胚葉は着床期胚のエピブラストと原始内胚葉の二つの細胞種に由来することがわかっていました。ところがシングルセル遺伝子発現データを一般的な解析ツールで細胞分化経路を追跡すると、エピブラストから内胚葉への繋がりも、原始内胚葉からの内胚葉に向けた分枝も消えてしまう。しかしRECODEで前処理をすると、この繋がりがクリアに浮かび上がりました」
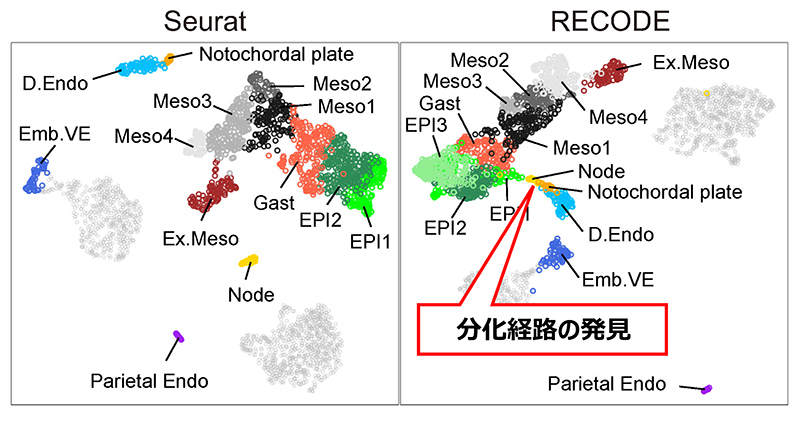 次元削減手法UMAPに基づく細胞分化経路解析の比較:一般的な解析法(Seurat)では変動の大きな遺伝子の選択と、PCAの主要主成分の選択による次元削減を行っている。マウス消化管は、エピブラスト由来の内胚葉と原始内胚葉由来の内胚葉の両者から発生することが知られているが、SeuratのUMAP解析では、エピブラスト(EPI)から内胚葉(D.Endo)に向けた分化経路や、原始内胚葉からの内胚葉に向けた分枝(Emb.VE)が消えてしまった。一方でRECODEでノイズを削減し次元を削減せずにUMAP解析を行うと、消えていた分化経路が正確に復元された。
次元削減手法UMAPに基づく細胞分化経路解析の比較:一般的な解析法(Seurat)では変動の大きな遺伝子の選択と、PCAの主要主成分の選択による次元削減を行っている。マウス消化管は、エピブラスト由来の内胚葉と原始内胚葉由来の内胚葉の両者から発生することが知られているが、SeuratのUMAP解析では、エピブラスト(EPI)から内胚葉(D.Endo)に向けた分化経路や、原始内胚葉からの内胚葉に向けた分枝(Emb.VE)が消えてしまった。一方でRECODEでノイズを削減し次元を削減せずにUMAP解析を行うと、消えていた分化経路が正確に復元された。
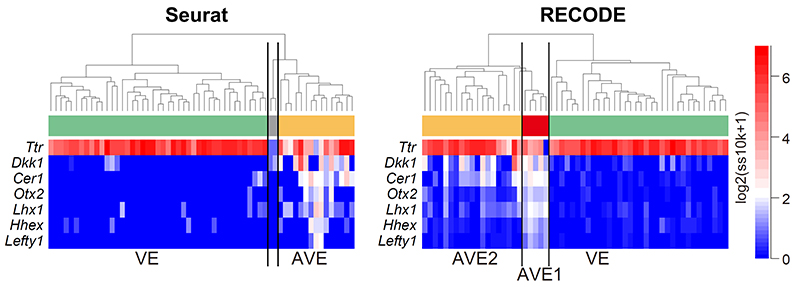 階層型クラスタリングによるAVE/VEの分類および主要遺伝子の発現分布:従来手法(Seurat)では全方臓腑側内胚葉(AVE)として分類されていた組織が、RECODEではAVE1とAVE2から構成されることを明らかにした。
階層型クラスタリングによるAVE/VEの分類および主要遺伝子の発現分布:従来手法(Seurat)では全方臓腑側内胚葉(AVE)として分類されていた組織が、RECODEではAVE1とAVE2から構成されることを明らかにした。
さらに体の前後軸の形成に関わる前方臓側内胚葉(AVE)のシングルセル遺伝子発現データをRECODEで前処理してからクラスタリングすると、意外なことが明らかになったという。
「AVEは高々10個程度の細胞からなる組織ですが、RECODEを使うと、AVEには二つの類似した、しかし確かに違う細胞種(AVE1とAVE2)から構成されることがわかりました。これまでの知見で知られていたAVEに特異的に発現する遺伝子すべてを発現していたのはAVE1で、AVE2では断片的で不均質な発現しか認められませんでした。このようにRECODEは新たな希少細胞種の発見を可能にします」
いずれもRECODEによりノイズが削減され、データ解析の解像度が上がったおかげで得られた結果だ。
「RECODEでシングルセル遺伝子発現データを前処理すれば、クラスタリングや統計量を用いた評価などあらゆるデータ解析結果を改善できます。シングルセルデータ解析の分野に良い貢献ができたと思っています」(井元さん)
WPI-ASHBiの研究者を中心にRECODEの利用者は少しずつ広がっているという。そこから希少細胞種や未知の細胞分化経路の発見や、がんなど遺伝子疾患メカニズムの解明の具体例が生まれれば、さらにRECODEが普及するだろう。
中村さんは今、RECODEを使ってカニクイザルをモデルに霊長類の初期発生を調べているという。
「ASHBiのメインテーマはヒト生物学で、私は着床直後の胚発生に興味を持っています。私の専門である発生学では古くからニワトリやマウスを使って研究が行われてきました。種を超えて保存される現象は多々ありますが、一細胞、全遺伝子レベルで解析が可能になった現代では、やはり鳥類やげっ歯類では種差が無視できなくなり、ヒトを知るには種差を排したよりヒトに近い生物学が求められています」
しかし倫理的な問題からヒトサンプルを手に入れることは不可能だ。
「そこで私はカニクイザルをモデルに、まずは着床期胚に存在する全細胞種を明らかにすべく、シングルセル解析を行っています。着床直後は、胚が本格的な形態形成と、均質で個性のないエピブラストから原始的な機能細胞(外/中/内胚葉)を創出する原腸陥入を開始させる根源的な時期であり、受精による命の始まりに対して”個”の始まりの時期であると考えています。着床直後のヒト胚発生の知見は約一世紀前に得られた形態学的なものしかありません。サル胚のシングルセル解析で得られた知見を基に、サルやヒトのES/iPS細胞なども駆使しつつ、ヒトの始まりにおける発生イベントを分子レベルで理解したいと思っています」(中村さん)
井元さんは遺伝子発現データの下流解析、特に時間的変化を捉える数学的手法の研究に取り組んでいるという。いつ、どんな遺伝子が発現して、細胞がどう変わるのか。時間軸に沿った解析の精度が上がれば、生物学の研究水準が底上げされる。
融合研究を妨げる「ノイズ」も削除
井元さんは中村さんと密に連絡を取れる環境があったから融合研究を前に進められたという。
「融合研究というのは互いになじみのない言葉、知見の歩み寄りから始まる手間のかかるものであり、さらに基本的には各々ほかにメインの研究もある。なので、せっかく研究が始まってもお互い忙しく、自然消滅することもよくあります。われわれの融合研究も気軽にコンタクトが取れる物理的な近さがなければ難しかったと思います。2019年に着任した頃、今WPI-ASHBiが入っているビルが完成してなくて、斎藤(通紀)研の部屋を貸してもらっていました。隣が中村さんの部屋だったので、何か分からないことがあるとすぐに聞きに行くことができました。廊下ですれ違ったときに話し合うこともよくあった。そのおかげで融合研究がスムーズに進んだのは間違いありません」
2020年に完成したWPI-ASHBiの施設は、数学系グループの研究室を取り囲むように、生物学系グループの居室や実験室が配置されている。生物学者と数学者の出会いのチャンスを増やすのが狙いだ。
「数学の新しい手法を紹介してもらったり、僕ら生物学者が数学的に解決できるかも?と思う問題を数学者に相談したりすることはよくあります。生物学者と数学者のマッチングです」(中村さん)
それぞれ専門を持つ研究者が異分野に関心を広げ、さらに融合研究に一歩踏みだして止まらず進み続けるには、研究者同士の物理的な距離の遠さ、専門知識の違いなど、いくつもの「ノイズ」がある。ノイズを取り払った先に、どんな融合研究が姿を現すだろうか。